12.5 Detection of DNA markers for disease genes or QTL's
Preceding page
Detection of DNA markers in general
The size of the entire genome in mammals and birds is about 3000 centi Morgan (cM) or recombination units.
This corresponds to that the largest chromosome is two to three hundred cM's long. The smallest ones are less than
100 cM's in length. There is a great deal of variation between the domestic animal species,
the mink has only 15
chromosome pairs and the dog has 39. There are also some differences in the recombination rate between males and
females, the females have on average 10 to 20 % higher frequency than the males. This is
particularly true for
the areas around the centromere.
Close linkage is easier to detect than loose linkage , and distances of more than 30 cM is in
practise impossible to detect, since it requires the typing
of a very large number of informative individuals (500-1000). On the other hand, if the distance is less than 10 cM
linkage can be detected with less than 50 informative gametes.
For a full coverage of the entire genome, there has to be an informative marker for each 20
cM. All in all this gives around 150 DNA markers to be typed for complete coverage.
For the marker to be of any use it has to be informative, this will on average be the case for about 50-70
% of them.
So to obtain a full set of markers in order to study a given family material, up to 300
evenly scattered markers have to be
available. Only the parent animal has to be pre-screened for heterozygosity for
all the 300 markers to get a useful
set consisting of 150 informative markers to complete a full analysis of the entire family.
Useful family material for detection of linkage to a disease gene
The segregation of a new recessive disease normally occurs by inbreeding at the ratio of 1 to 3. The statistically most
optimal cituation is for a dominantly inherited disease with a segregation of 1 to 1.
For recessive diseases a 1 to 1 ratio
can only be obtained by classifying offspring in a back cross.
To localize a recessive disease gene is needed more than 20 offspring, where 10 of which have the
disease and the carriers parent animals.
The
analysis can start by marker 1 on chromosome 1 and performing a statistical test
by continuing like that with a new marker until linkage is found. Linkage can be found
in the first marker, but it can also be found in the last marker, number 150. On average only
half of the markers (75) have to be applied before linkage is found. A
statistically significant association would appear as follows:
diseased not diseased
Genotype ------------------------------------
aa | 10 3 | 13
Not aa | 4 13 | 17
------------------------------------
| 14 16 | 30
Here there are only of few recombinants, 3, with the genotype aa which
not diseased, and 4 being diseased and not having the genotype aa.
To summarize: To identify a DNA marker for a disease gene animal material
from a few related families should exist,
comprising more than 20 offspring of which at least 10 should have the disease. For species where there exist a marker chip,
used this of course as it is both faster and cheaper, you can also do with fewer animals because the chip has
several markers for each centi Morgan, so it can be expected that there are markers which have no recombinants.
When linkage has been found, it is natural to continue using markers between the two markers
providing the linkage. The final
goal will always be to identify the real disease gene. When linkage has been found, comparative studies can also be
initiated. Candidate genes for the disease might be found by looking at the corresponding chromosome areas in other
species, which are already known. An alternative to the classic marker analysis might be
a careful study of the disease and thereby finding a candidate gene from another
species. A candidate gene is a gene with a fair chance of causing the disease when comparing the aetiology of the disease. If one or more candidate genes exist, the analysis starts
by typing these. If it is the right gene, complete association
is found.
DNA markers applied for studies of quantitative trait loci
(QTL) demand far more data
material than it takes for finding a marker for a disease gene, if the gene are
to be detected in a normal outbred
population. QTL's correspond to the loci, which are discussed in the definition of breeding value in
chapter 6. The
most distinct problems in the detections of a QTL are that it is unknown whether
or not a QTL segregates in a given family. If it segregates how big is the effect on a given
trait? Another problem arises if a
QTL is detected. How is it possible to discern the hypothesis that one or two genes cause the QTL
effect? Planning of a QTL study has to be very carefully done in order to
obtain any sensible results. A large amount of animals have to be
typed in order to estimate a given QTL with sufficient precision. It is important that
a number of characteristics are measured for the animals to be typed.
Here all possible production traits can be mentioned, for example diseases and other easily observable traits. The optimal number of
informative gene markers is around 150 with a
distance of about 20 cM, as mentioned. An alternative to the study of
QTL's in normal populations can be
the study of F2
individuals from special exotic crosses, as for instance between domestic swine and
wild boar. The detected QTL's in such studies cannot
be applied in connection with the normal breeding, but they can be used for pointing
out candidate genes. The variation
between F2 animals from exotic crosses can be very large. Therefore,
to find a certain number of QTL's the number of animals to be typed
is smaller than in a normal outbred population.
Use of grand sire or sire design for detecting QTL's. The two classic designs, the grand sire and the sire
designs are shown in Figure 12.2.
Figure 12.2. Showing the sire and the grand sire designs for detecting QTL's. With test of offspring for marker allele and
phenotype measurements. The contrasts can be evaluated by classic statistical
tests.
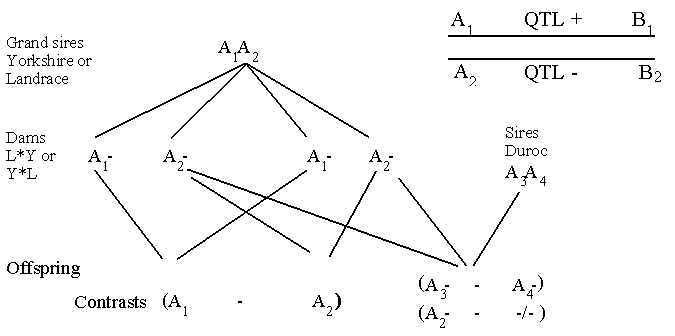
The grand sire is heterozygote with respect to the genotype A1A2. Half of his
sons will receive the A1 allele and
the other half the A2 allele. Now a contrast can be made between the average breeding value of those sons having
received the A1 and those having received the A2 allele. In the grand sire design the
classification of genotypes is only done in the
sons, whereas the phenotype data derives from the grand daughters. This design has especially been used
in the estimation of QTL's in dairy cattle.
In the sire design the genotype and the phenotype data come from the same animals. As can be
seen from Figure 12.2, more contrast can be estimated than in the grand sire design. In the sire design the major
problem is that a large number of animals has to be typed. For instance,
to detect a specific least statistically significant difference in
disease frequency, when the average disease frequency is 10 %, the number
given in the table below has to be typed. The binomial variance and a t-test contrast have been used for an approximation.
No with A3 No with A4 SD least statistically significant difference
5000 5000 .006 1.8 % units
500 500 .019 5.7 % units
100 100 .042 12.6 % units
For 10 % units difference to be detected at least 200 animals have to be classified. As
might be known, there are many causes for variation in the occurrence of disease and an average disease frequency of 10
% is fairly high. If the disease
frequency is lower more animals is needed to detect a given QTL.
Next page