12.5 Detección de marcadores de ADN de los genes de la enfermedad o de QTL
Página anterior
Detección de marcadores de ADN en general.
Detección de marcadores de ADN en general. El tamaño del genoma completo en mamíferos y
aves es de unos 3000 centi Morgan (cM) o unidades de recombinación. Esto corresponde al
que el cromosoma más grande es de largo de dos a tres cientos cM. Los más pequeños son
menos de 100 cM longitud.
Existe una gran variación entre las especies de animales domésticos, el visón tiene sólo
15 pares de cromosomas y el perro tiene 39. También hay algunas diferencias en la tasa de
recombinación entre machos y hembras, las hembras tienen en promedio 10 al 20
porciento mayor frecuencia que los machos. Esto es particularmente cierto para las zonas en
torno a la centrómero.
Para ligamiento estrecha es más fácil detectar que el ligamiento suelto y distancias de más de
35 cM es en la práctica imposible de detectar, ya que requiere le uso de un gran
número de individuos informativos (500-1000). En la otra parte, si la distancia es inferior
a 10 cM ligamiento puede ser detectada con menos de 50 gametos informativos.
Para una cobertura del genoma completo, tiene que haber un marcador informativo para
cada 20 cM. En todo esto da alrededor de 150 marcadores de ADN para tener una cobertura
completa.
Para que el marcador de cualquier uso tenga que ser informativo, esto será en promedio el caso
alrededor del 50-70 porciento de ellos. Así que para obtener un conjunto completo de marcadores
para estudiar un determinado material familiar, hasta 300 marcadores uniformemente dispersos deban
ser disponibles. Sólo el parental tiene que ser preseleccionados para heterocigotos para todos
los 300 marcadores obtener un útil conjunto compuesto por 150 marcadores informativos para completar
un análisis completo de todas las familias.
Material de familia útil para la detección de ligamiento a un gen de la enfermedad.
La segregación de una nueva enfermedad recesiva normalmente se produce por consanguinidad en la proporción de 1 a 3.
El situación estadísticamente más óptimo es una enfermedad hereditaria es con una segregación de 1 a 1.
Para localizar una enfermedad recesiva es necesaria investigar al menos 20 descendencias, 10 de los cuales
tienen la enfermedad y los hermanos son normales. El análisis puede empezar por marcador 1 cromosoma
1 y realizar una prueba estadística por continuar así con un nuevo marcador hasta que encuentra el ligamiento.
Ligamiento puede encontrarse en el primer marcador, pero también se puede encontrar en el último marcador,
número 150, En promedio, sólo la mitad de los marcadores (75) debe aplicarse antes de que se encuentre el ligamiento.
Una asociación estadísticamente significativa aparecería como sigue:
Enfermo no enfermo
Genotipo ------------------------------------
aa | 10 3 | 13
No aa | 4 13 | 17
------------------------------------
| 14 16 | 30
Aquí hay sólo de algunos recombinantes, 3, con el genotipo aa que no enfermas y 4 está enfermo
que no tienen el genotipo aa.
En resumen:
Para identificar un útil marcador de ADN es necesario que exista un material de animales en
unas pocas familias relacionadas donde el gen de enfermedad segrega
con mas de 20 animales de los cuales al menos 10 debería tener la enfermedad.
Cuando se ha encontrado el ligamiento, es natural para seguir utilizando los marcadores entre los
dos marcadores proporcionando el ligamiento. El objetivo final será siempre identificar el gen
de la enfermedad real. Cuando se ha encontrado el ligamiento, también se pueden iniciar estudios
comparativos. Genes que podrían utilizarse para la enfermedad podrían encontrarse mirando en las
áreas de cromosoma correspondiente en otras especies, que ya son conocidos. Una alternativa para
el análisis de marcador clásico podría ser un cuidadoso estudio de la enfermedad y encontrar así
un gen de candidato de otra especie. Un gen de candidato es un gen con una oportunidad justa de
ser causante de la enfermedad cuando se comparan la etiología de la enfermedad. Si existen uno o
varios genes de candidato, el análisis se inicia investigando estas. Si es el gen de la derecha,
se encuentra una asociación completa.
Marcadores de ADN cuando se aplican para estudios de loci de un rasgo cuantitativos (QTL, ingles
quantitative trait loci )
demanda mucho más datos que toma para encontrar un marcador para un gen de la enfermedad.
QTL corresponde a los loci, que se han discutido en
la definición de valor en el capítulo 6 de valor de cría. Los problemas más distintos en las
detecciones de un QTL son que se desconoce o no segrega un QTL en el seno de una familia determinada.
¿Si segrega cuán grande es el efecto en un rasgo dado? Otro problema se plantea si se detecta un QTL.
¿Cómo es posible discernir la hipótesis de que uno o dos genes causan el efecto de la QTL?
Planificación de un estudio QTL ha de hacerse muy cuidadosamente sin el fin de obtener
ningún resultado razonable. Una gran cantidad de animales debe mediado con el fin de
estimar un determinado QTL con suficiente precisión. Es importante que una serie de características
se mide a los animales. Aquí todos los rasgos de producción posible pueden ser mencionados,
por ejemplo enfermedades y otros rasgos fácilmente observables. El número óptimo de marcadores de
genes informativo es alrededor de 150 con una distancia de aproximadamente 20 cM, como se ha mencionado.
Una alternativa para el estudio de QTL en poblaciones normales puede ser el estudio de individuos de F2
de cruces exóticas especiales, como por ejemplo entre animales domésticos de la especie porcina y el cerdo.
El QTL detectado en esos estudios no puede aplicarse en relación con la cría normal, pero se pueden utilizar
para señalar de genes que podrían. La variación entre animales de F2 de cruces exóticas puede ser muy grande.
Por lo tanto encontrar a un cierto número de QTL el número de animales es menor que en
una población normal.
Uso de gran padre o padre diseño para la detección de QTL en el cerdo. Los dos diseños clásicos,
el gran padre y los diseños de padre se muestran en la Figura 12.2.
Figura 12.2. Mostrando el padre y el gran padre diseños para la detección de QTL. Con la prueba de descendencia para mediciones de alelo y fenotipo de marcador. Los contrastes pueden evaluarse por pruebas estadísticas clásicas.
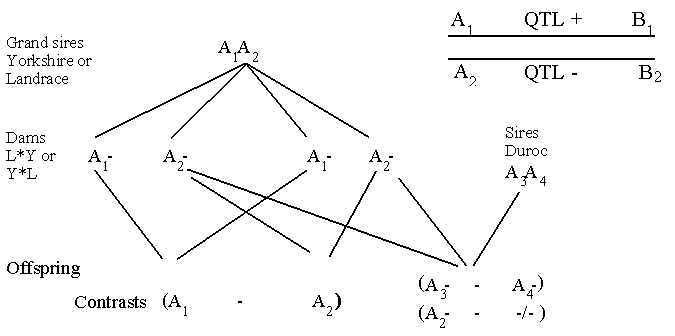
El gran padre es heterocigoto con respecto al genotipo A1A2.
La mitad de sus hijos recibirán la A1 y la otra mitad el A2 alelo. Ahora puede
hacer un contraste entre
la media en el valor de esos hijos de haber recibido la A1 y esos el
alelo A2 de cría. En el diseño de gran padre la clasificación de los genotipos sólo
se realiza en los hijos, mientras que los datos del fenotipo derivan de las
nietas. Este diseño ha sido utilizado especialmente en la estimación de QTL en
vacas lecheras.
En el padre diseñar el genotipo y los fenotipos datos provienen de los animales de los mismos.
Como puede verse en la Figura de 12,2, más contraste se puede estimar que en el diseño de gran padre.
En el padre diseño el gran problema es que un gran número de animales deben ser utilizados. Por ejemplo,
para detectar una diferencia estadísticamente significativa menos específica en la frecuencia de la
enfermedad, cuando la frecuencia de la enfermedad promedio es de 10 porciento, el número dado en la
tabla siguiente debe utilizar. La variación binomial y un contraste de t-prueba han sido utilizados
para una aproximación.
No con A3 No con A4 SD el menos diferencia estadísticamente significativa
5000 5000 ,006 1,8 porciento unidades
500 500 ,019 5,7 porciento unidades
100 100 ,042 12,6 porciento unidades
Para 10 porciento unidades de diferencia para ser detectado al menos 200 animales tiene que ser clasificados. Como podría ser conocido, hay muchas causas para la variación en la aparición de la enfermedad y una frecuencia de enfermedad promedio del 10 porciento es bastante alta. Si la frecuencia de la enfermedad es menor más animales es necesaria para detectar un QTL determinado.
Página siguiente