Påvisning af DNA-markør generelt
Hos pattedyr og fugle er størrelsen af det samlede genom ca. 3000
centi Morgan (cM) eller rekombinationsenheder, der er dog en betydelig variation
mellem arter. De største kromosomer er to til tre hundrede cM lange,
mens de mindste er under 100 cM lange. Her er der også betydelig forskel
mellem de enkelte arter, hvor mink kun har 15 kromosompar og hunden
har 39. Hos pattedyr er der forskel på rekombinationshyppigheden mellem
hanner og hunner, det er således, at hunnerne har højere (10 to 20 %)
rekombinationshyppighed end hannerne, hvilket især gør sig gældende i områderne
omkring centromerene.
Tæt kobling er lettere at påvise end løs kobling,
afstande over 30 cM er i praksis næsten umulige at
påvise idet det kræver et meget stort antal informative
individer (500-1000). Det
er således, at kobling indenfor 10 cM kan påvises på grundlag af under
50 informative stk afkom fra en analysekrydsning.
For at man kan dække genomet regnes der med, at der
skal anvendes en DNA-markør for hver 20 cM, dette giver
ialt 150 DNA-markører, der skal types for at dække
hele genomet.
For at DNA-markøren kan anvendes skal den være informativ,
dette er for de fleste DNA-markører kun tilfældet i
50-70 procent af familierne, der skal analyseres. For at
få et komplet set af DNA-markører, må det derfor prescreenes
hos forældredyrene med op til 300 markører, for at få
et brugbart sæt på 150 til analyse af en given familie.
Egnet familiemateriale til påvisning af sygdomsgen
Udspaltning af et sjældent forekommende recessivt
sygdomsgen sker normalt i forbindelse med indavl
i forholdet 1 til 3. Den statistisk mest optimale situation
er en 1 til 1 udspaltning, men det kan normalt ikke skaffes
ved recessive sygdomsgener, men man kan nøjes med at type
lige så mange raske individer som syge for at få de
mest optimale analyse betingelser.
For at finder frem til
sygdomsgenet skal man regne med typning af ca 10 forældre dyr
der er anlægsbærere mere end 20 stk afkom, hvoraf de 10 har
sygdommen. Man starter med typning af markør 1, derefter
udføres en statistisk analyse. Man fortsætter med typninger af
det næste markørgen indtil man finder koblingen.
Man kan finde den koblede allel ved først DNA-markør,
eller man kan finde den ved markør nr 150,
men man skal i gennemsnit anvende 75 markører før man når frem
til det koblede gen. Den statistisk signifikante
association vil da se således ud:
syg ikke syg
Genotype ------------------------------------
AA | 10 3 | 13
ikke AA | 4 13 | 17
------------------------------------
| 14 16 | 30
DNA-markører anvendt til QTL studier
DNA-markører for quantitative trait loci (QTL) kræver
langt større datamaterialer end for at finde en DNA-markør
for et sygdomsgen, såfremt genet skal påvises i en normalt udavlet
population. QTL'er svarer til de loci, der er omtalt
under definitionen af avlsværdi i lektion 6. De
fremherskende problemer ved påvisning af et QTL er endvidere, at
man ikke ved om der er et der spalter ud i en given
familie. Og man ved heller ikke på nogen måde, hvor stor
effekten i givet fald skal være på en given egenskab.
Der er yderligere det problem, at når man først har
påvist en QTL, kan man ikke på nogen måde være sikker på, at
effekten skyldes et gen, da man ikke umiddelbart kan skelne den fra
to koblede gener med hver sin effekt på den givne egenskab.
Planlægning
af et QTL studier er en sag der skal gøres meget
omhyggeligt, for at man kan forvente at nå frem til
et fornuftigt resultat.
Da man skal type mange dyr for at estimerer en given
QTL tilstrækkeligt præcis til at den kan få praktisk betydning,
og der bør gennemføres statistisk power beregninger for at finde
frem til hvor mange dyr der skal types for at finde en given effekt.
Det er tillige vigtigt at typningerne kan anvendes til at evaluere så
mange egenskaber som muligt. Så når man starter med
at planlægge et QTL studie er det vigtigt at få målt
så mange egenskaber som muligt på de pågældende dyr.
Her kan nævnes alle mulige mål for produktion, mål
for sygdomsforekomst og andre observerbare egenskaber.
Antallet af DNA-markører, der
skal anvendes, svarer dog til de ca. 150, som blev nævnt under
afsnittet om påvisning af DNA-markør for sygdomsgen.
Alternativt til studier af QTL'er i normalpopulationer
kan være at anvende studie af F2 individer
fra specielle eksotiske krydsninger som feks. mellem
tamsvin og vildsvin. Sådanne fundne QTL'er vil imidlertid
ikke umiddelbart kunne anvendes i forbindelse
med det normale avlsarbejde. Sådanne påviste QTL'er
kan imidlertid anvendes i forbindelse med udpegning af kandidatgener,
der kan bidrage til en dybere forståelse af de almindelige
fysiologiske funktioner.
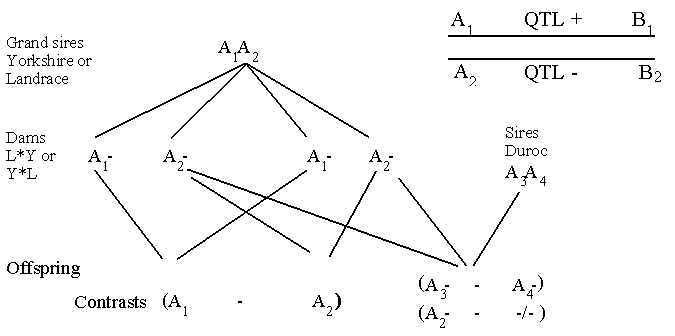
Sire og grandsire design til påvisning af QTL'er
Grand sire designet har været anvendt en hel del indenfor
studie af kvægpopulationer. Det gå ud på at man kun
typer tyrene, idet man udvælger tyre der har et større
antal sønner. Man opdeler da sønnerne i to klasser
dem der har fået et bestemt gen fra faderen og dem der har
fået det andet. Man kan så danne en statistisk kontrast
mellem middel avlsværdien af de to klasser, dem med eller
uden genet fra grand sire. Dette kan gøres for alle
DNA-markører og der er pt. fundet frem til QTL'er for
fedt % og mælkeydelse i amerikanske bestande af Holstein
Frisian, men QTL'erne har endnu ikke været anvendt i praktisk
avlsarbejde.
Fordelen ved grand sire designet er, at der skal types
forholdsvis få individer og man har en 1 til 1 udspaltning blandt
afkommet, men en af begrænsningerne er,
at der kun kan estimeres additive effekter, hvilket er
tilfældet i sire designet.
Sire designet; her types alle individer der måles såvel
som forældre dyrene. Men da DNA-markørerne er mest interessante
for egenskaber med lav heritabilitet skal der types mange
dyr for at få et rimeligt præcist skøn for effekterne. Man
får her alle tre genotype klasser blandt afkom med 1:2:1 udspaltning.
Variansen på en geneffekt (middelværdi) er omvendt
proportional med antallet, på hvilket geneffekten
er baseret.
En kontrast mellem to effekter kan i princippet
evalueres med
et t-test, her er det også den middelværdi der er baseret
på færrest antal der bestemmer hvor stærk testet er.
Nedenstående er vist en tabel med mindste signifikante forskelle. Forskellene er påvist i en population med middel sygdomsfrekvens på 10 %. Syg ikke syg er binominalfordelt med variansen 0,1*0,9/N hvor antallet er testede dyr i en klasse. S.D. er beregnet som sqrt(0,1*0,9/N + 0,1*0,9/N) og t = dif/S.D. og anvendelse af 5% signifikansgrænser.
| Antal A1 | Antal A2 | S.D. | Mindste signifikante differens i sygdomsfrekvens |
| 5000 | 5000 | 0,00600 | 1,8 % enheder |
| 500 | 500 | 0,0190 | 5,7 % enheder |
| 100 | 100 | 0,0420 | 12,6 % enheder |
| Figur 12.2. Viser de to klassiske design sire og grand sire til påvisning af QTL'er med test af afkom for markørgen og fænotype måling. Contrasterne kan sammenlignes med t-test.
|